
Integrations¶
The files written by GECCO are standard TSV and GenBank files, so they should be easy to use in downstream analyses. However, some common use-cases are already covered to reduce the need for custom scripts.
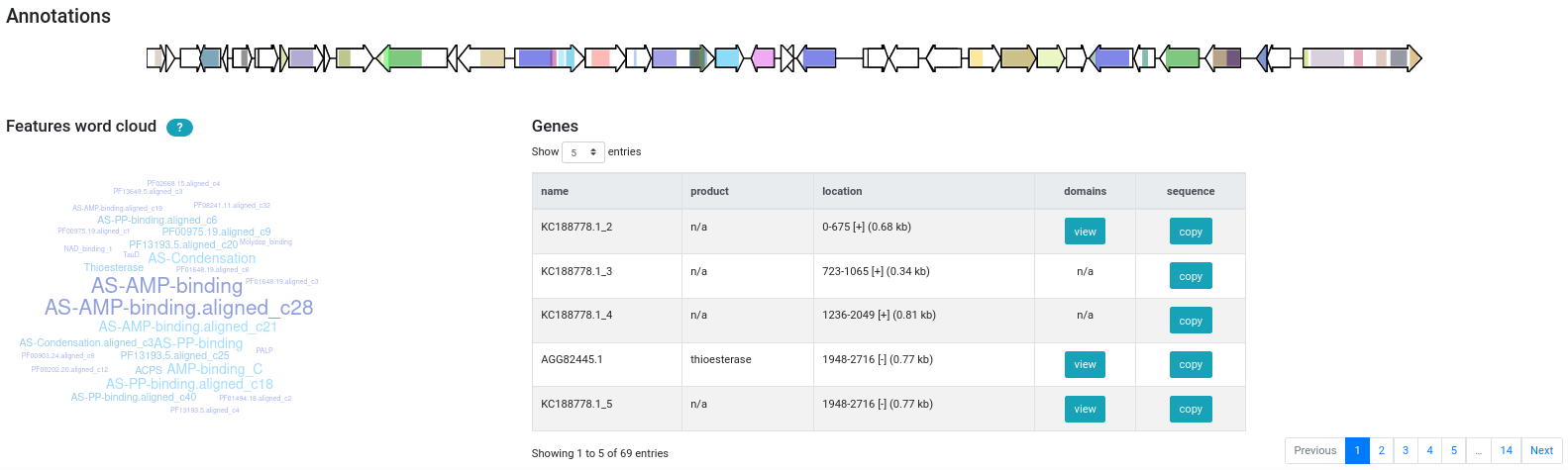
Genome Feature¶
GECCO outputs tables containing the location of BGCs in TSV format to retain
as much metadata as possible for each predicted BGC. These tables are easy to
manipulate with a library such as pandas
or polars. However, most biology visualization
tools can load arbitrary features in GFF format,
which may be used to visualize predicted BGCs across an entire genome.
Since v0.9.7 GECCO offers the option to generate a GFF file from its output:
$ gecco run -g KY646191.1.gbk -o output_dir
$ gecco convert clusters -i output_dir --format gff
The output folder will contain an additional GFF file with each cluster as a single feature:
$ tree output_dir
output_dir
├── KC188778.1_cluster_1.gbk
├── KC188778.1.clusters.gff
├── KC188778.1.clusters.tsv
└── KC188778.1.features.tsv
Feature Coloring¶
Starting from v0.9.6, GECCO will attempt to color the gene features in the
output GenBank files based on their molecular function. GenBank has no
standard way of doing so, but many software offer their own way of coloring
features. At the moment, GECCO outputs color qualifiers that should be
supported by APE,
EasyFig,
Benchling or
SnapGene as shown below:
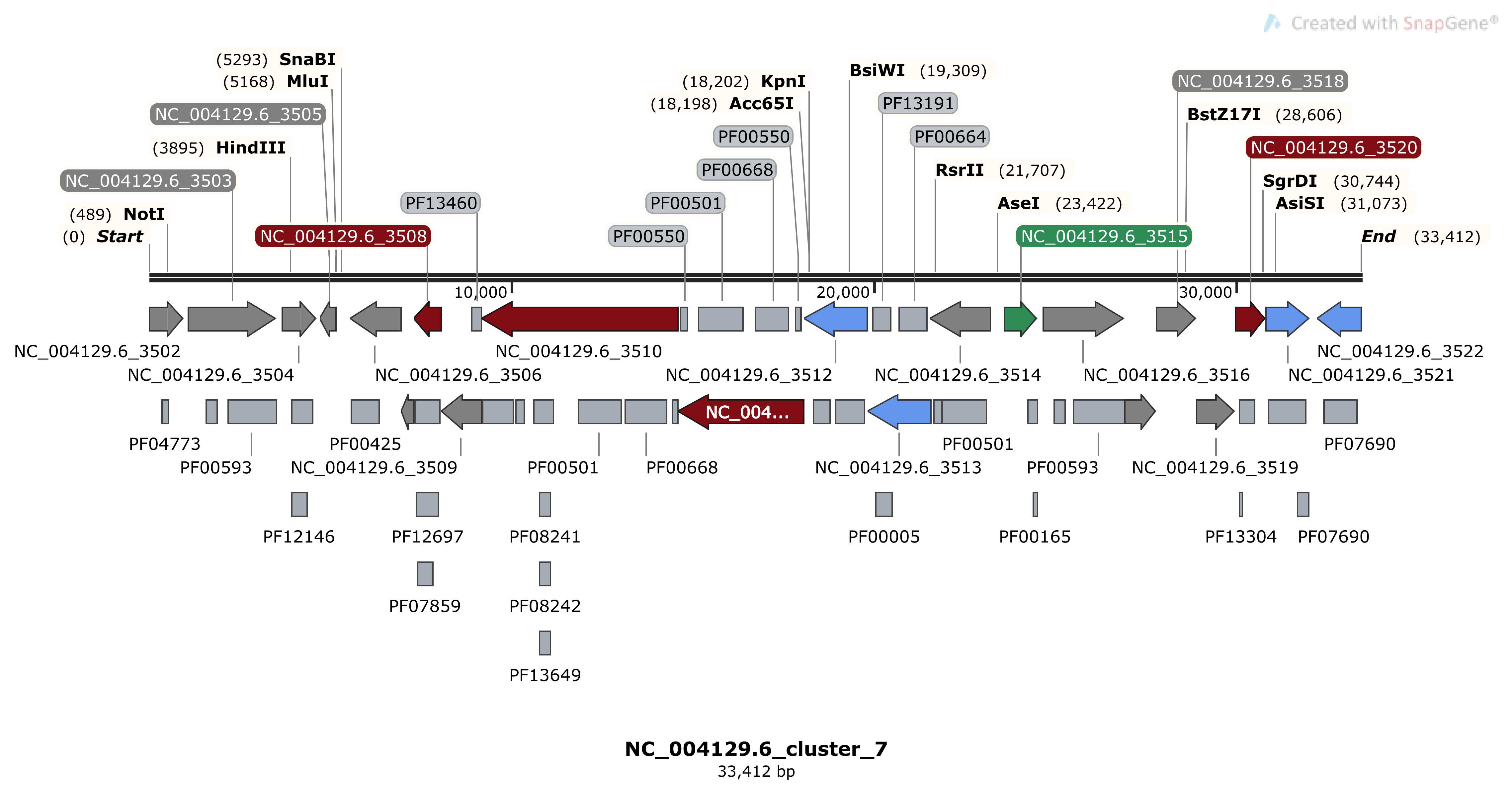
The color code is the same as MIBiG, green for regulatory proteins, blue for transporters, red for biosynthetic proteins, and grey for unknown function protein.
AntiSMASH¶
Since v0.7.0, GECCO can natively output JSON files that can be loaded into
the AntiSMASH viewer as external annotations. To do so, simply run
your analysis with the --antismash-sideload option to generate an
additional file:
$ gecco run -g KC188778.1.gbk -o output_dir --antismash-sideload
The output folder will contain an additional JSON file compared to usual runs:
$ tree output_dir
output_dir
├── KC188778.1_cluster_1.gbk
├── KC188778.1.clusters.tsv
├── KC188778.1.features.tsv
└── KC188778.1.sideload.json
0 directories, 4 files
That JSON file can be loaded into the AntiSMASH result viewer. Check
Upload extra annotations, and upload the *.sideload.json file:
When AntiSMASH is done processing your sequences, the Web viewer will display BGCs found by GECCO as subregions next to the AntiSMASH clusters.
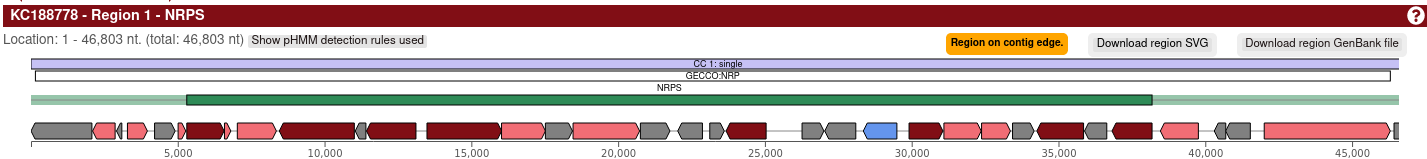
GECCO-specific metadata (such as the probability of the predicted type) and
configuration (recording the --threshold and --cds values passed to
the gecco run command) can be seen in the dedicated GECCO tab.

BiG-SLiCE¶
GECCO outputs GenBank files that only contain standard features, but BiG-SLiCE requires additional metadata to load BGCs for analysis.
Since v0.7.0, the gecco convert subcommand can convert GenBank files
obtained with a typical GECCO run into files than can be loaded by BiG-SLiCE.
Just run the command after gecco run using the same folder as the input:
$ gecco run -g KY646191.1.gbk -o bigslice_dir/dataset_1/KY646191.1/
$ gecco convert gbk -i bigslice_dir/dataset_1/KY646191.1/ --format bigslice
This will create a new region file for each GenBank file, which will be detected by BiG-SLiCE. Provided you organised the folders in the appropriate structure, it should look like this:
$ tree bigslice_dir
bigslice_dir
├── dataset_1
│ └── KC188778.1
│ ├── KC188778.1_cluster_1.gbk
│ ├── KC188778.1.clusters.tsv
│ ├── KC188778.1.features.tsv
│ └── KC188778.1.region1.gbk
├── datasets.tsv
└── taxonomy
└── dataset_1_taxonomy.tsv
3 directories, 6 files
BiG-SLiCE will be able to load and render the BGCs found by GECCO:
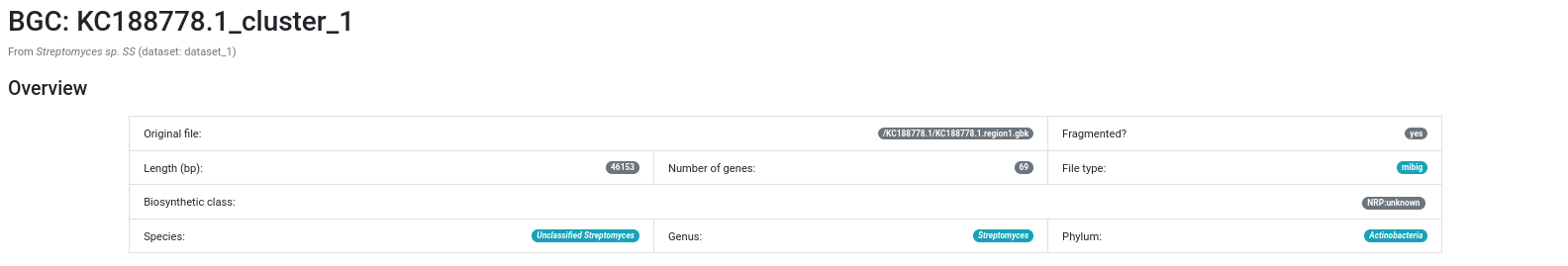
Warning
Because of the way BiG-SLiCE loads BGCs coming from GECCO, they are always marked as being fragmented.